Data retrieval: sequences
In order to study microbial processes in the selected environments, we need to know who is there, what can they do, and what are they doing. The way of addressing these questions is by using DNA sequencing techniques. There are two main approaches to retrieve sequences.
a) Microbial cells are first isolated either in pure culture or by cell sorting. Then, their nucleic acids can be sequenced.
Pure cultures
 The genome of a pure culture can be sequenced directly. The different DNA pieces (reads) have to be assembled into larger fragments (contigs and scaffolds). Eventually, the whole genome can be put together in a single closed circle. Genome recovery is usually very good, but many of the abundant bacteria in nature are difficult to isolate in pure culture.
The genome of a pure culture can be sequenced directly. The different DNA pieces (reads) have to be assembled into larger fragments (contigs and scaffolds). Eventually, the whole genome can be put together in a single closed circle. Genome recovery is usually very good, but many of the abundant bacteria in nature are difficult to isolate in pure culture.
Single sorted cells
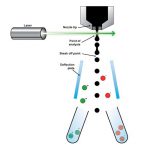
Each cell is sorted into a well in a plate. The cells are lysed and their genome amplified before it can be sequenced. Amplification is not uniform and usually only a part of the genome can be obtained. The advantage is that it belongs to a naturally abundant bacterium. These genomes are called SAGs (single amplified genomes).
b) Nucleic acid (DNA or RNA) molecules are retrieved directly from the ecosystem.
Next, a particular gene can be selected for study, for example the 16S rDNA gene. In this case an amplification step is needed with primers specific for the gene and the result is a collection of amplicons, all of the same length and containing just the desired gene. This approach is very useful for studies of diversity, to know ‘who is there.’
Another approach is to sequence the whole mixture of nucleic acids. This provides the metagenome (when DNA is sequenced) or the metatranscriptome (when the mRNA is used). One advantage is that the usually biased amplification step is avoided. But the main advantage is that we can study the function of the genes in the community. The genes present (metagenome) and the genes being expressed (metatranscriptome) become available to study what capabilities do the microorganisms have, and which genes are being expressed at each time.

Data processing: cleaning, assembly and annotation
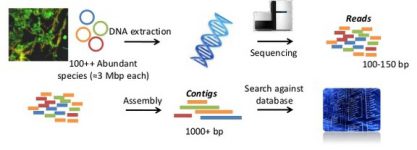
Millions of tiny DNA sequences. That is what we get when we sequence the nucleic acids. These small bits (reads) are each part of the complete genome of their original species. The bioinformatics analysis attempts to reconstruct as much as possible of these genomes, find the genes coded in it and provide hypotheses for the function of each of these genes.
Cleaning
Even though current high throughput sequencing techniques have low percent of errors, they provide millions of sequences and, thus, there will be plenty of sequencing errors. The first step involves identifying and rejecting the sequences with errors. Often, sequences need to be trimmed in order to get rid of the ends where errors accumulate. Another problem is the appearance of chimeras, when partial sequences from two different organisms are artificially combined by the sequencing process. These need to be removed from the data set.

Picture from Mads Albertsen
Assembly
The next step in the analysis is assembling, that is, joining together these small sequences (reads) to recover longer pieces of the genome (contigs). If an organism is very dominant in the ecosystem or there are few species in it (such as in extreme environments) or we have sequenced a lot of DNA, we can have enough metagenomic reads to reconstruct the full genome. Otherwise, we will have smaller contigs (this is how we call the fragmentary pieces of a genome product of an assembly) that can nevertheless represent much of the genome of the species. In a metagenomic assembly, usually we will have thousands of contigs corresponding to pieces of the genomes of the species in the sample.
Annotation
Finally, we want to identify the genes encoded in our contigs, because the genes are elemental units coding for proteins that will perform one (or more) functions. The genes are the basic units for describing the functioning of an organism. We use gene prediction tools for knowing which genes are encoded in a piece of DNA like a single contig. Then, we can compare them with the millions of other genes that are already known to us and are stored in huge databases. This is known as homology searching. A close similarity between genes means that it is likely that the new one is doing a similar function to the already known one (of course one needs to be very careful when defining what is «close similarity», but you get the idea). Also, a close similarity between genes will inform about their putative origin as well (its taxonomy). Thus, homology searching allows annotation of the genes both functionally and taxonomically.
Knowing the functions encoded in the genes and their taxonomy, we can produce a picture of the possible functioning of the full microbial community and the role of each species.
Binning
The metagenome contains, in theory, all the genes from all the microorganisms in the sample. Once sequenced and assembled, the contigs can be binned into taxonomically uniform bins (similar to species or strains). Often, these bins can be used to reconstruct fairly complete genomes (MAGs, metagenome assembled genomes). These belong to abundant bacteria in nature and do not require the amplification step of SAGs. However, binning may not always work sufficiently well for the organisms of interest.
Data analysis: How to use sequences to study microbial ecology
We can use the information on the presence and functional capabilities of the organisms in the ecosystem to obtain ecological information on the possible associations between organisms, or the particular role they are fulfilling in the ecosystem.
For inferring associations, we can look for concerted variations in the presence or abundances of each species in different samples. Co-variation between species is often an indicator of a direct or indirect association between them. We use generalized Lotka-Volterra models (an extension for n species of the classical predator-prey models of classical ecology) to detect these significant co-variations.
The correlations between species in different samples are often complex and involve more than one species. Therefore, it is useful to organize these associations in co-occurrence networks. Linked to the ecological information of the samples in which the species co-occur, this analysis can inform on the conditions in which a particular association is taking place. This informs us on the possible ecological or biochemical basis of the putative association
A detailed study of the role of a species in the ecosystem is obtained by creating models that simulate the functioning of the cell. To that end, we use the functions encoded in the genome of an organism to create a metabolic map informing of all the metabolic reactions in the cell. With it, we can infer how the cell will use the available metabolites, which ones will produce, and the possible metabolic interactions with other species via the complementation of their needs.
